Maladies rares: à chaque syndrome sa classe de mutations
Les récents progrès en matière de séquençage du génome humain ont permis de découvrir la cause de nombreuses maladies rares. Le professeur Alexandre Reymond, directeur du Centre intégratif de génomique (CIG) de l’UNIL, en collaboration avec une équipe mondiale de généticiens, décrit dans l’édition du 28 janvier 2021 de l’«American Journal of Human Genetics» comment trois classes de mutations, situées sur un même gène, engendrent trois syndromes neurodéveloppementaux spécifiques.
Identifier la ou les mutation(s) génétique(s) à l’origine d’une maladie parmi les 3,2 milliards de lettres qui constituent l’alphabet de notre génome représentait, il y a encore peu de temps, un réel défi. L’arrivée de nouveaux outils d’analyse, notamment le séquençage à haut débit, a permis de déterminer l’enchaînement des lettres pour un fragment d’ADN donné à des coûts et dans des temps fortement réduits. «Nous pouvons désormais définir de manière précise les régions responsables d’une maladie, diagnostiquer cette dernière et trouver potentiellement de nouveaux traitements», relève Alexandre Reymond, professeur ordinaire à la Faculté de biologie et de médecine et directeur du Centre intégratif de génomique de l’UNIL.
Un réseau international de généticiens
Par l’entremise de collaborations qui regroupent des généticiens et des cliniciens du monde entier, il est dorénavant possible de trouver la cause de maladies extrêmement rares, dites «orphelines». C’est le cas notamment des trois pathologies qui font aujourd’hui l’objet d’une publication dans l’American Journal of Human Genetics (AJHG). Codirigée par Alexandre Reymond, cette étude a été menée en partenariat avec les chercheurs néerlandais Lisenka Vissers (Radboud University) et Simon Fisher (Max Planck Institute for Psycholinguistics).
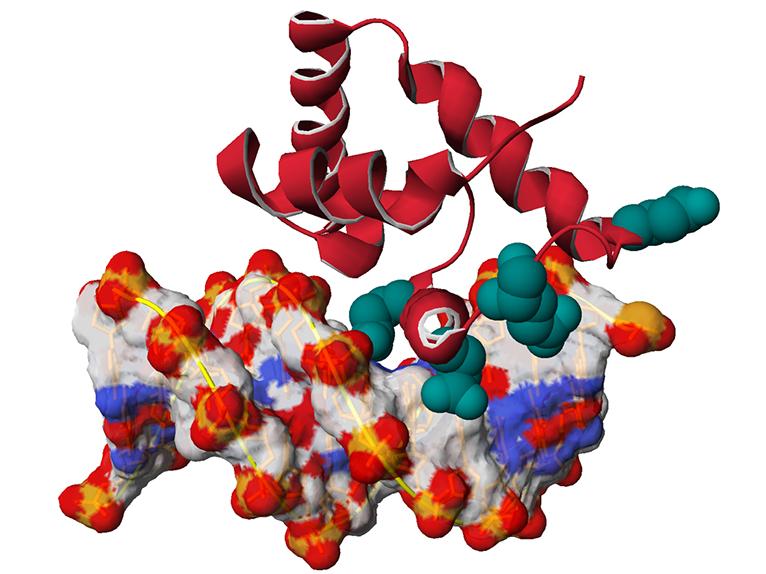
La mise en commun de données provenant de généticiens spécialisés dans les maladies humaines, répartis sur quatre continents et dans douze pays, a permis d’identifier 42 patients présentant des mutations sur un gène spécifique appelé SATB1. Ce dernier renferme l'information nécessaire à la fabrication d'une protéine qui peut se lier à l’ADN, avec pour effet de diminuer l’expression d’autres gènes.
Trois classes de mutations
Les mutations mises en lumière par les scientifiques peuvent être regroupées en trois classes différentes. La première, présente chez huit patients, contient des mutations qui détruisent la fonction du gène SATB1, avec pour résultat une inhibition de la moitié de la production de la protéine. La seconde rassemble quatre mutations qui engendrent la production d’une forme plus courte et par conséquent moins performante de la protéine, en raison du mauvais positionnement de cette dernière dans la cellule. La troisième classe, enfin, regroupe les mutations identifiées chez les trente patients restants. Celles-ci transforment la protéine et la rendent plus active. La protéine ainsi modifiée «colle» davantage à l’ADN et diminue d’autant l’expression des gènes qu’elle régule.
Des troubles bien définis
Les chercheurs ont découvert que ces trois modes d’action différents étaient associés à trois syndromes neurodéveloppementaux distincts. «Les protéines qui fonctionnent "trop bien" causent une déficience intellectuelle sévère souvent accompagnée d’épilepsie, d’un trouble de l'articulation de la parole (dysarthrie) et d’un faciès spécifique. Le manque de protéines est, pour sa part, associé à une cognition diminuée mais plus proche de la normale, des problèmes au niveau de la vision et un faciès particulier. Finalement, les patients ayant des mutations qui produisent des protéines raccourcies et mal localisées présentent un syndrome intermédiaire», détaille Alexandre Reymond.
Aller au-delà du simple séquençage
Ces résultats démontrent que toutes les mutations ne sont pas égales et qu’il est primordial de comprendre leur mode d’action pour élucider la cause des maladies génétiques. «Il faut aller plus loin que le simple séquençage, qui n’est qu’une première étape», conclut le professeur lausannois.
par Manuela Palma De Figueiredo - Communication FBM